
Facility: Comparing sample preparation techniques
When comparing sample preparation methods you need to consider what success looks like. BioMS’s goals for sample prep methods are as follows:
Ease of use: The method must be comparatively easy to follow as this means it is more likely to be used and less likely to go wrong.
Robust and reliable: Following on from ease of use the methods should ideally work first time and every time as much as possible.
Scaleable: Projects in the facility range from a couple of samples to 100+ and so our methods must be able to cope with sample numbers in that scale. With that in mind we much prefer plate based methods, especially those that have the potential to be automated.
High recovery/precise: The higher the recovery of peptides the less variability is seen in the results, this is especially important in the label free quantification that is used by most BioMS users. High sensitivity is essential for high sensitivity methods and high precision essential for determining subtle differences.
Clean: The methods must get rid of contaminants that effect the LC-MS system. Examples of the effects we need to remove include blocking columns (expensive to replace), changing the peak elution positions (you get worse quantification results), dirty the mass spec (results in big delays for system cleaning) and general drops in sensitivity (any of the above and many other ‘sneaky’ causes that are hard to track down).
Quick: Methods should be reasonably quick o perform to ensure a rapid throughput. Definitely less than 2 days and preferably less than 1 day.
Affordable: Mass spec experiments can scale quickly and are frequently not originally costed in grants so we need costs as low as possible to encourage usage.
There are many mass spec/proteomics sample prep methods available and we’ll assess some of the most popular ones below:
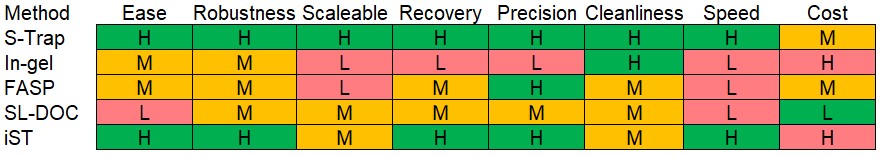
S-Traps: S-Traps work by trapping solvent precipitated proteins on a quartz frit held in a custom device or plate. Previous experiments in BioMS looking at trapping precipitated proteins indicate that the mechanism of S-Traps is probably more complex than simple precipitation and probably involves some form of HILIC interaction as well. The trapped proteins are then given a solvent wash and this wash can be customised for tricky samples that contain lots of lipids such as brain and heart. The digestion is rapid, taking just an hour with a high enzyme to substrate ratio. the rapid digestion results in an increased amount of missed cleaved peptides however the digests are highly reproducible and can be extended if desired. Overall we have found that the combination of Covaris AFA disruption with S-Trap processing and our R3 based plate sample clean up is highly effective. It produces high quality data sets on a wide range of samples from mammalian tissues to plant tissues and microbes and with high sensitivity, being regularly used for laser capture dissected material. Samples are extremely clean resulting in a massive reduction in the contamination of the MS systems and from there less failed runs. This procedure should be used as a default for all samples.
In-gel digestion: In-gel digestion is the longest running method in the facility, being available as a service since the facility was created. We initially did a lot of ‘gel walks’ or gel pixilation where whole lanes are cut up into small sections and are analysed separately but this has mostly been replaced by ‘gel-tops’ where all the sample is loaded into the top 5mm of a gel and analysed as a single sample. It is still the best way of looking at a single protein in a complex background and is especially useful for protein characterisation. It works well for removing lipids and detergents as proteins are trapped in polyacrylamide and washed with solvents however recoveries can be low and variable as the process depends upon trypsin entering the gel pieces and this can be limited by the size of the gel pieces produced as trypsin would have further to travel in large gel pieces. PAGE gels produce many different size pieces when they are cut – they look like they shatter when examined with a microscope. Different size distributions will have different digestion efficiencies
FASP: Filter Aided Sample Preparation (FASP) uses ultrafiltration devices to concentrate, buffer exchange and digest proteins and it is one of the widest used proteomics sample prep techniques. We use it predominately for looking at cell culture media samples in secretomics as concentration is built into the process. We don’t use it much for proteomics anymore as the process is lengthy due to the centrifugation steps, it has trouble with many detergents other than SDS, we’ve had a number of samples fail due to the centrifugation units and it is hard to do the process at scale due to the fiddliness of the tubes and limited capacity in centrifuges.
SL-DOC: The sodium laurate (SL) with deoxycholate (DOC) methodology was developed in the facility to address the issues seen in FASP and in-gel digestion. It uses mild detergents SL and DOC to solubilise proteins and allow in solution digestion followed by a two-phase solvent extraction to remove the detergents and many lipids. The method can work well but it can also be tricky to use and doesn’t work with some types of samples and so has been superseded by S-Trap methods.
iST: The iST digestion kits have been developed by Preomics, a company that has spun out of Matthaeus Mann’s lab. They are very well regarded and have been adopted by major facilities such as those in the Crick Institute. BioMS have tried the kit as part of the GFP-Trap immunoprecipitation protocol from Chromatek and they worked well. Our issue with the kit are that they are expensive and our Covaris/S-Trap/R3 pipeline is working very robustly so there is no drive to change.
Tags: #Facility, sample prep, proteomics





0 Comments